Highlights of our Work
2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
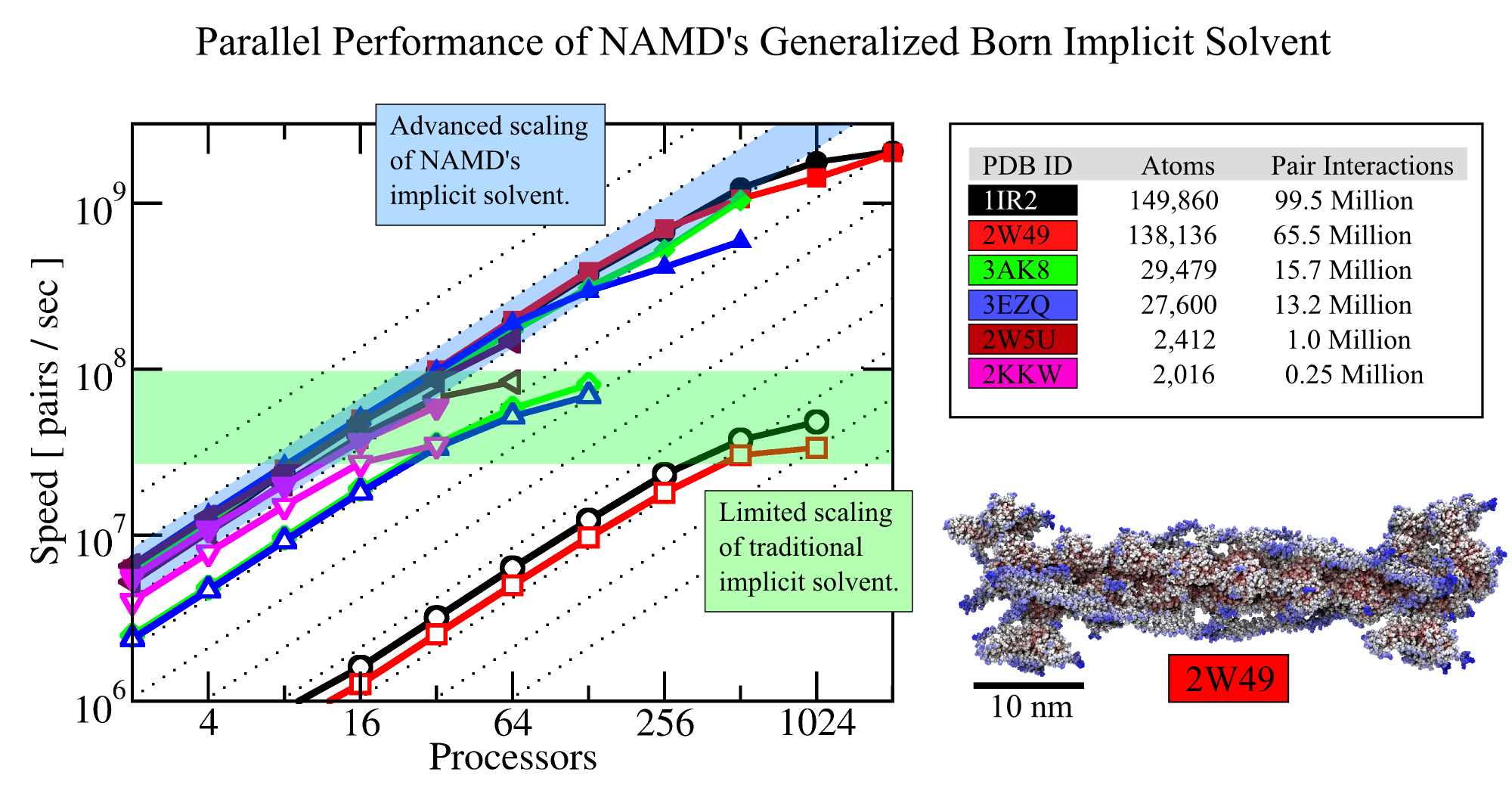
image size:
573.0KB
Olga Svinarski and VMD
Water is ubiquitous; living cells and the cells' biomolecules are greatly influenced by water.
Accounting for water in computational modeling, a key access route to imaging cell processes, comes at a stiff price: up to 90% of the simulation volume needs to be a water bath embedding simulated biomolecular machinery. The sheer number of water molecules arising here, sometimes millions, and the strong hydrodynamic drag resisting biomolecular dynamics, slows down simulations tremendously. Computational biologists had invented a means to replace water by a kind of continuous ether, so-called implicit solvent, that accounts for key electrostatic features of water around biomolecules, but does not slow down simulations. Computational hinderance through explicit water is getting worse with increasing size of biomolecular machines, but so far the implicit solvent description could not be effectively employed to model large systems using supercomputers. In a recent study, the situation has been repaired and the use of an implicit solvent in molecular dynamics simulations of systems as large as the whole ribosome (containing 300,000 ribosome atoms and eliminating 2,700,000 water atoms) have been documented. In a further study, the implicit solvent description has been extended to account also for hydrophobic interactions at biomolecular surfaces; the extension took advantage of delegating calculations both to GPU and CPU as the latter is more suitable for the evaluation of surface interactions. More information regarding NAMD's parallel computer-ready implicit solvent can be obtained from the NAMD website.
Nov
2005 and
Threading DNA electrically through nanometer-sized pores, so-called
nanopores,
holds promise for detecting and sequencing DNA (see Nov
2005 and Oct
2004
highlights).
Nanopore measurements tend to be the more sensitive the smaller the pores are.
The material graphene, which is just one atom thick and looks like a
two-dimensional ``honeycomb" made up of carbon atoms, offers the ultimate
physical resolution for measuring DNA (the stacking distance between
base-pairs in DNA is about 0.35 nm). As reported recently, molecular dynamics
simulations using NAMD revealed the motion of DNA being threaded through
graphene nanopores at atomic level resolution. Simulations not only agree
qualitatively with previous experiments on DNA translocation through graphene
nanopores, but go one step further than the experiments and suggest how
individual base pairs can be discriminated. The recent computational study is
one further example for the guidance
that molecular dynamics simulations provide in nanosensor development (see a
recent review).
More
information can be found on our graphene
nanopore website.
Computer simulations of the biomolecular processes in human cells guide
better understanding of health and disease as well as development of
dietary supplements and pharmacological treatments. Such simulations are
extremely demanding and, in fact, all too often still limited by
technological feasibility. However, technological advances are being
brought to bear on computer simulations in biomedicine through highly
dedicated biomedical engineers, who have often speerheaded uses of new
computer technologies such that they became available in biomedicine much
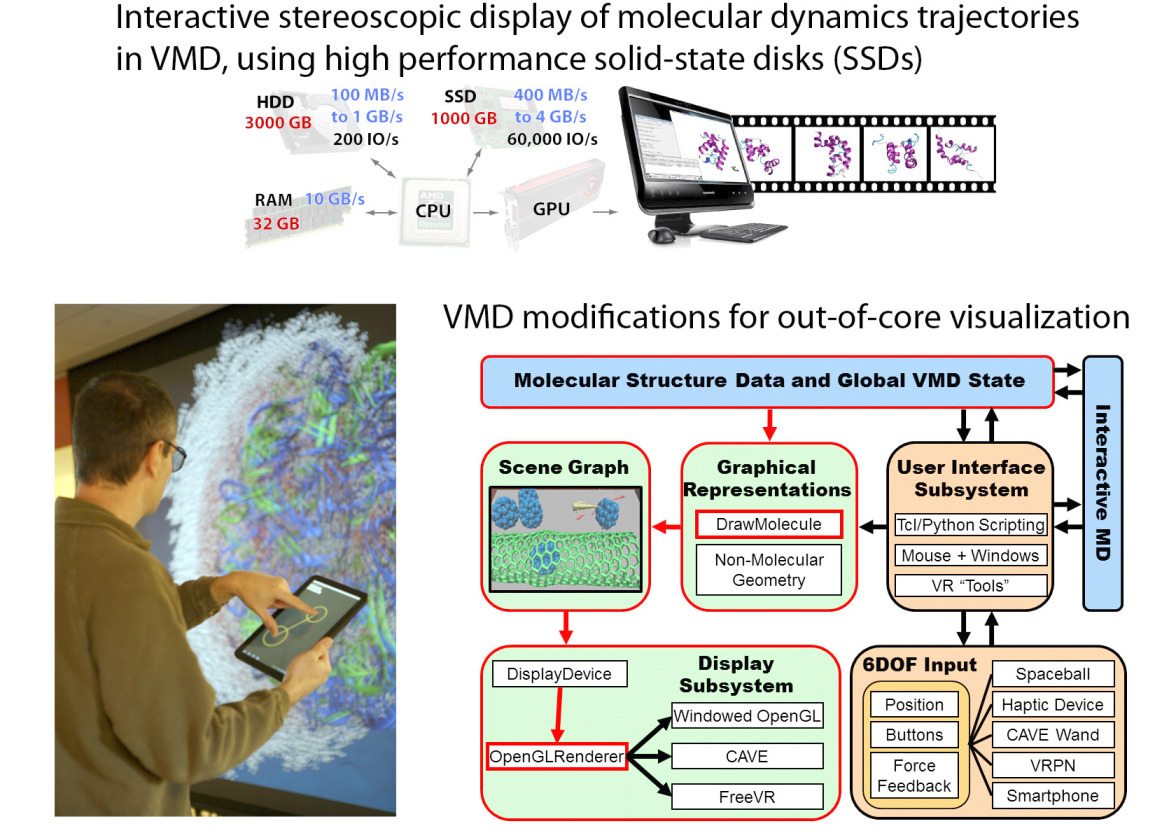
sooner than in other fields. A case in point is solid state disk (SSD)
technology that can serve as extremely fast and large computer memory.
Conventional RAM (random access memory) is fast, but limited in size
due to cost; the well known hard disks (HDs) can hold large data sets at
an affordable price, but are slow. The new SSDs are in the middle ground,
faster than HDs, slower than RAM, offering large data storage at an
affordable price. Modern uses of SSDs in smart phones and tablets attest
to the usefulness of SSDs. The biomolecular visualization and analysis
software VMD in its next release (VMD 1.9.1) makes the power of SSDs as a
huge, yet fast storage medium available to biomedical researchers.
As reported,
this will allow them to view and analyze through VMD on the fly
Gigabytes-to-Terabytes of simulation data, that are being raked into a
computer at the rate of up to 4 Gigabytes per second (one high definition video
of a long movie per second!). For more on this and other revolutionary
features of VMD 1.9.1 see our VMD web site.
NAMD molecular dynamics program excels at
simulating, in atomic detail, the complex molecular machinery of living cells.
NAMD now enables ..." />
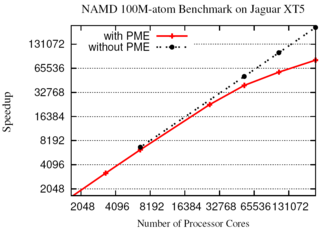
The NAMD molecular dynamics program excels at
simulating, in atomic detail, the complex molecular machinery of living cells.
NAMD now enables hundred-million-atom simulations using the
full capabilities of the nation's fastest supercomputers
due to parallel programming innovations reported in
a paper
at the SC11 supercomputing conference.
While parallel supercomputers have massive amounts of memory in aggregate,
any part of the machine can directly access only a small fraction.
To fit the molecular blueprint of cellular structures such as the
chromatophore in the memory
of such machines, a compressed data
format was developed that exploits the repetitive nature of proteins,
nucleic acids, lipid membranes, and solvent.
This format requires only a small amount of data to be copied to all parts
of the supercomputer, while the remainder is read from disks in parallel and
distributed across the machine.
Simulation output and the balancing of work loads are similarly distributed.
The Charm++
parallel programming system, on which NAMD is built, was also extended
to allow the molecular structure and other data to be shared among the
increasing number of cores in modern processors.
A hundred-million-atom simulation can now run on supercomputers
with only 2 gigabytes of memory per node, such as the IBM BlueGene/P.
The enhancements are available to computational biologists world-wide in
special memory-optimized
versions of NAMD 2.8.
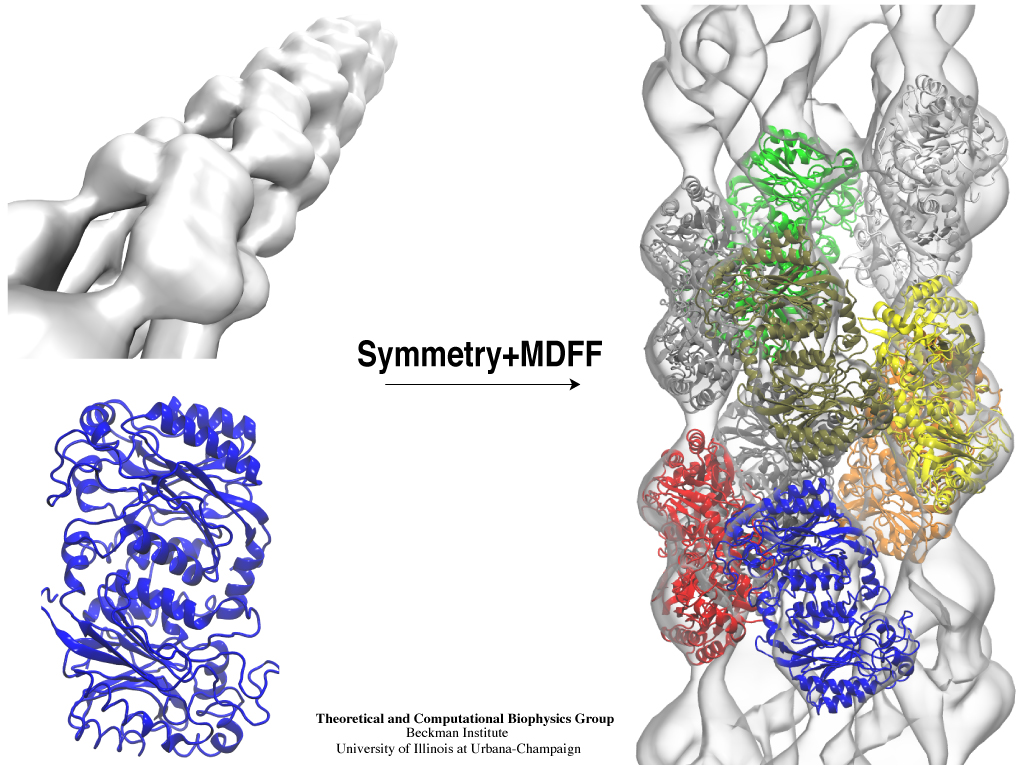
Inorganic nature brings about symmetry that one can admire,
for example when one sees a polished diamond.
Living nature, too, brings about symmetric structures;
for example, many processes in living cells are carried out
by highly symmetric protein complexes. In case of living cells
symmetry comes about partially out of physical or geometrical necessity,
but also partially out of its usefulness for the intended purpose.
Hence, understanding symmetry in biology goes beyond related studies
of the beauty of symmetry in the physical and mathematical sciences
as one also asks: Does symmetry help?
Put it another way: Symmetry in living cells is beautiful and useful!
The proteins in symmetric complexes are often imaged by electron microscopy (EM),
but unfortunately not at a resolution high enough to recognize chemical detail,
as is needed in most cases, e.g., in case of the study of a symmetric multi-enzyme system.
Computational biologists have developed a method to solve the resolution problem,
using high resolution images obtained through X-ray scattering of related molecules and
matching them through molecular dynamics using
NAMD
to the image seen in EM. This method, called molecular dynamics flexible fitting (MDFF) has been
highlighted
previously and is described on our
MDFF website.
As reported recently,
MDFF has now been extended to determine the atomic-level structure of symmetric multi-protein systems.
MDFF has been applied successfully to three highly symmetric multi-protein systems:
(i) GroEL-GroES, a protein complex that assists proteins to fold properly into their native conformation;
(ii) a nitrilase multi-enzyme system that converts massive amounts of molecules into forms
more suitable for a bacterial cell;
(iii) Mm-cpn, a protein complex supposedly involved in assisting protein folding
in so-called archaebacteria.
In all cases the symmetry of the protein complexes plays a key role.
For more details, see the
Method
section of our
MDFF website.
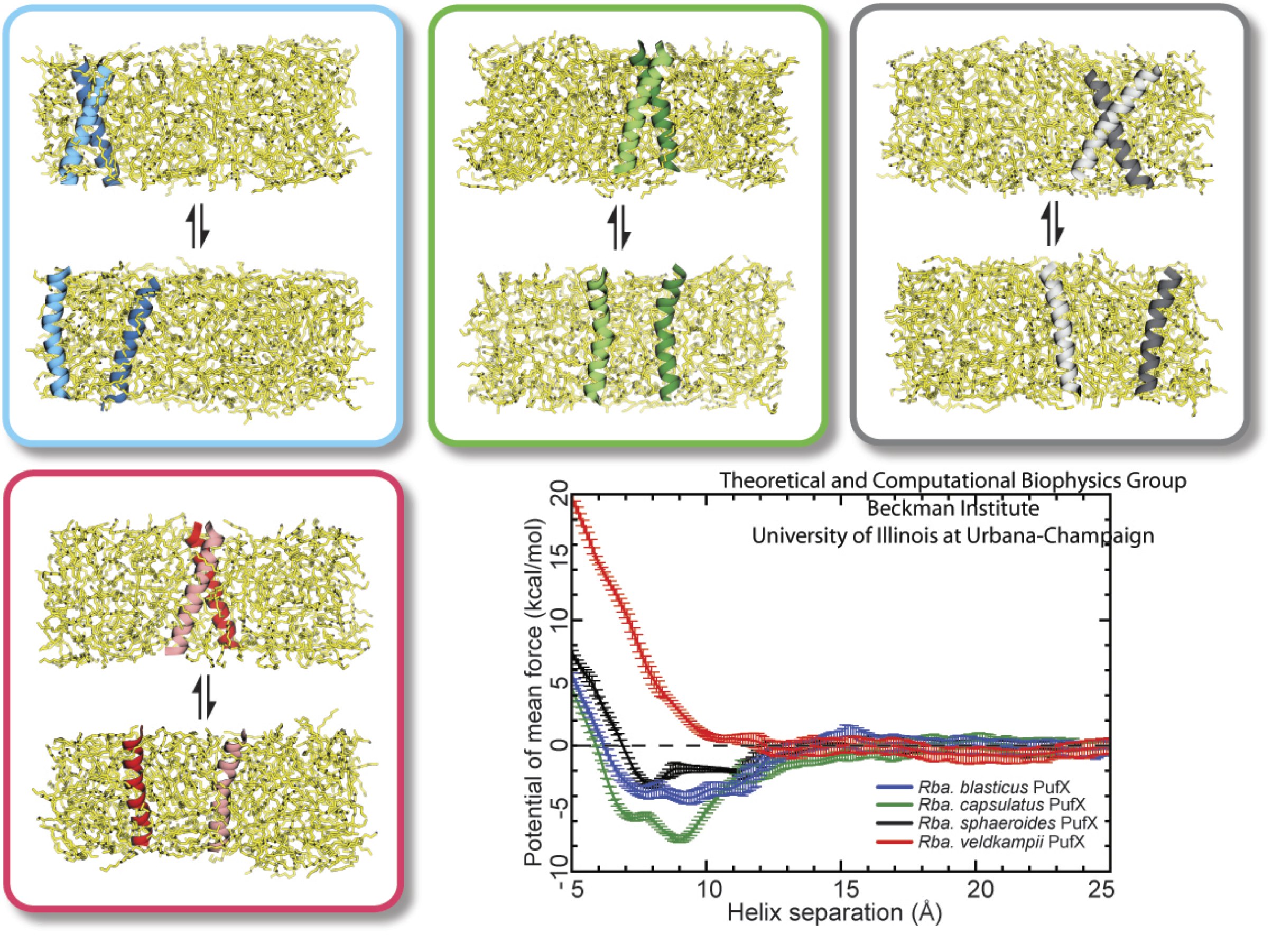
image size:
284.7KB
made with VMD
Bacteria contain the simplest photosynthetic machineries found in nature. Higher organisms like algae and plants practice photosynthesis in a more elaborate but principally similar manner as bacteria. But even for its simplicity, the bacterial photosynthetic unit is not without its unsolved mysteries. Take, for example, the crucial photosynthetic core complex, which performs light absorption and the initial processing of the light energy. In certain bacterial species, the core complex are each ring-shaped singlets, but in some other bacterias they can be 8-shaped doublets, formed by the association of two ring-shaped core complexes. The question arises: What holds doublets together. It turned out that doublets arise when bacteria bacterial contain two copies, i.e., twins, of an additional small protein called PufX. Recently, computational biologists and biochemists have come together to study PufX from different bacteria. They found an interesting trend showing that bacterial species with PufX that associates with another PufX protein also contain core complexes that form 8-shaped doublets. On the other hand, bacteria whose PufX is incapable of associating with another PufX have only singlets of ring-shaped core complexes. These new results resonate with a proposed role of PufX as the protein that holds two core complexes together to form 8-shaped doublets. Bacteria with PufX that cannot perform this role therefore only have singlets of ring-shaped core complexes. The needed simulations were done with NAMD. More details can be found on our photosynthetic core complex website and in a previous highlight on PufX.
The genes of organisms, like plants and animals, offer the blueprint, not only to build the organism anew from a seed or fertilized egg cell, but also to adapt the living organism to its habitat and life experience. For example, a type of tree
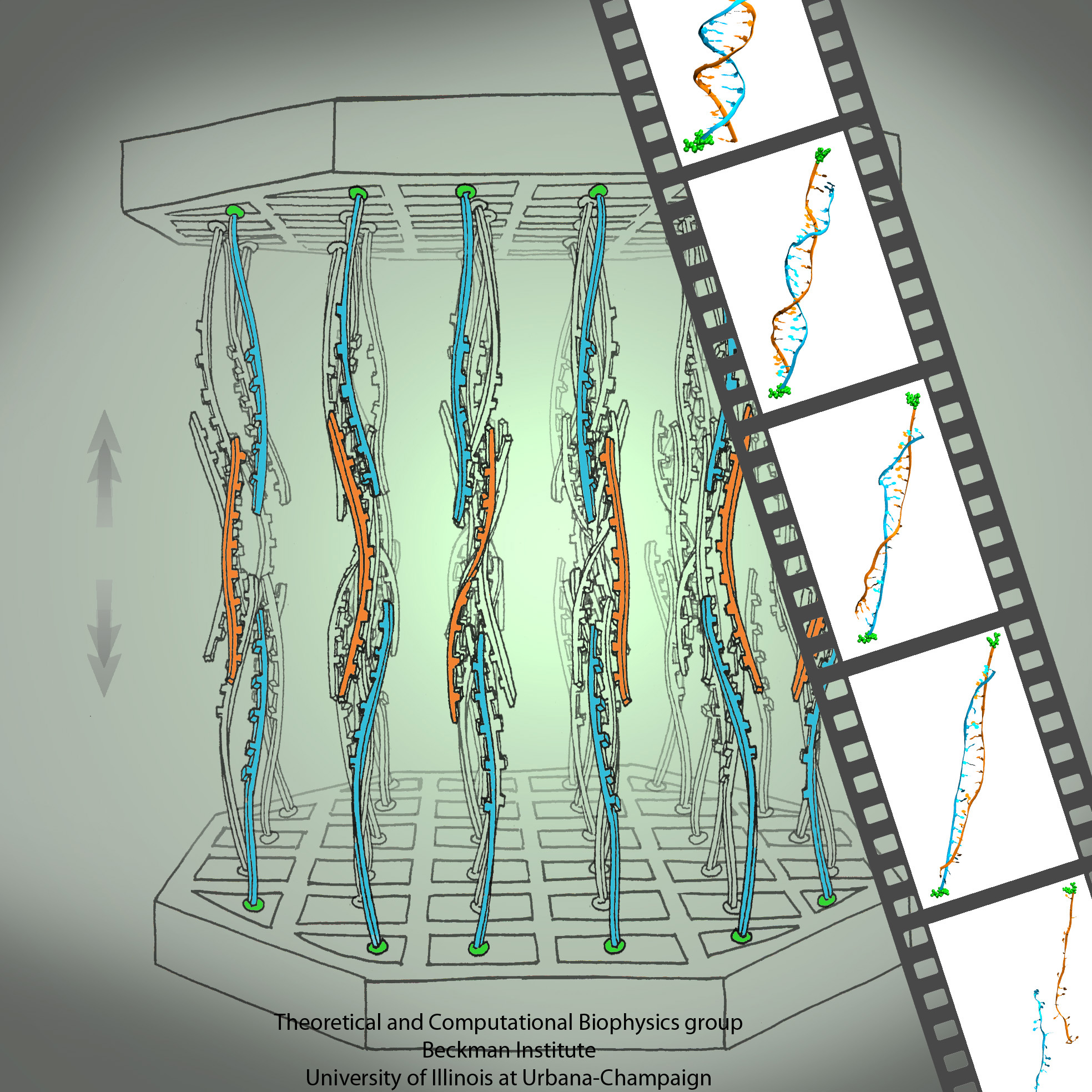
growing in an arid or wet region will adapt expression of its genes for root growth optimal to circumstances. A child living on a scarce or abundant diet or with little or much physical activity will adapt body growth accordingly. The needed, life time adaptation requires an individual's change in gene expression. This change is the subject of epigenetics. One control element in epigenetics is that cytosine bases of an organism's DNA become methylated in a chemical reaction in which a hydrogen atom is replaced by a methyl group (CH3). Another control element involves hydroxymethylation of cytosine bases where the H atom is replaced by a hydroxymethyl group (CH2OH); hydroxymethylation arises mainly in brain tissue. DNA methylation and hydroxymethylation patterns depend on an organism's individual history; aberrant patterns can be the cause of diseases, for example, of certain cancers. It is known that the proteins involved in gene expression can recognize methylated sites of DNA and, thereby, direct gene expression; DNA methylation also affects the packing of DNA in the chromosomes. However, methylation and hydroxymethylation may also affect gene expression directly; indeed, experiment and computational modeling with NAMD suggest this as an intriguing third way for methylation and hydroxymethylation to regulate gene expression. Methylation is shown, as reported recently, to make it more difficult to separate the two strands of DNA, as is necessary during gene expression. An earlier experimental-computational study had revealed already that methylated DNA can pass narrow synthetic nanopores more readily than unmethylated DNA can (see the Feb 2009 highlight). A recent experimental-computational study has shown that also in the case of hydroxymethylation mechanical properties of DNA become altered, namely, the forces needed to separate the two strands of DNA are strongly affected. The three experimental-computational findings advance our understanding of methylation and hydroxymethylation-based epigenetics and of how our body adapts to our life style and environment. More on our DNA methylation and hydroxymethylation website.
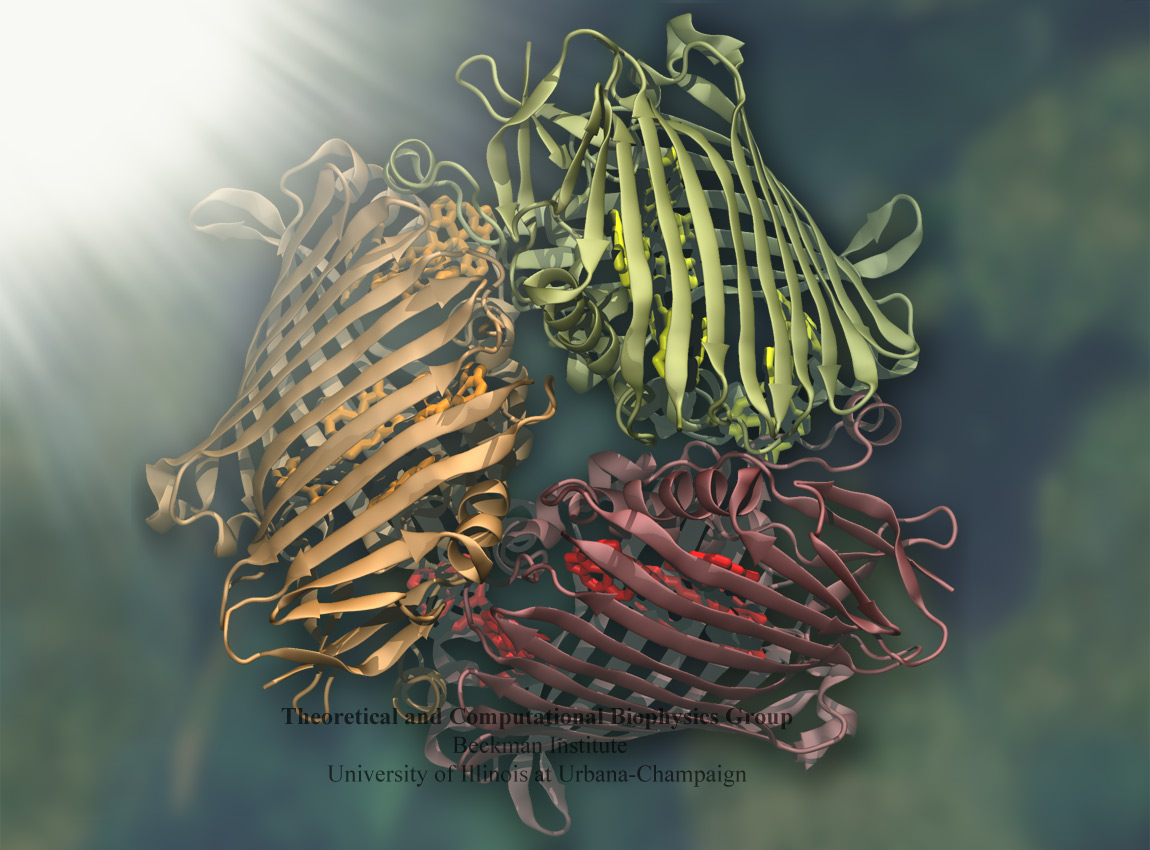
image size:
218.3KB
made with VMD
Green sulfur bacteria are life forms that use sun light as a main food
source. They
harvest the light by absorbing the sun's photons with their chlorosome
system, an assembly of
thousands of chlorophyll molecules. Absorption produces electronic
excitation of the chlorophyll molecules, but the excitation is only
short-lived (lifetime of about 1 nanosecond) and needs to be turned
quickly enough into a
more stable form of energy. The latter is achieved in a protein complex
called the reaction center, but for this purpose the
electronic excitation needs to travel from the chlorosome to the
reaction
center through a protein complex called the Fenna-Matthews-Olson (FMO)
protein. FMO is a crucial bottleneck, acting as an
energy faucet, through which the short-lived excitations need to
flow in a fraction of a nanosecond. Electronic
excitations are quantum phenomena highly sensitive to thermal noise.
Biophysicists spend much effort
to measure thermal effects on FMO electronic excitation flow. Now
researchers have complemented measurements through calculations and
have shown why FMO function is actually robust against thermal noise.
Using a
combination of classical and quantum mechanical calculations they
quantified the thermal noise present in FMO (reported recently
here and
here), and
determined that thermal noise greatly reduces quantum
coherent excitation in the transport through FMO (reported
here), but that
does not seem to be detrimental to
excitation flow. More information can be found here.
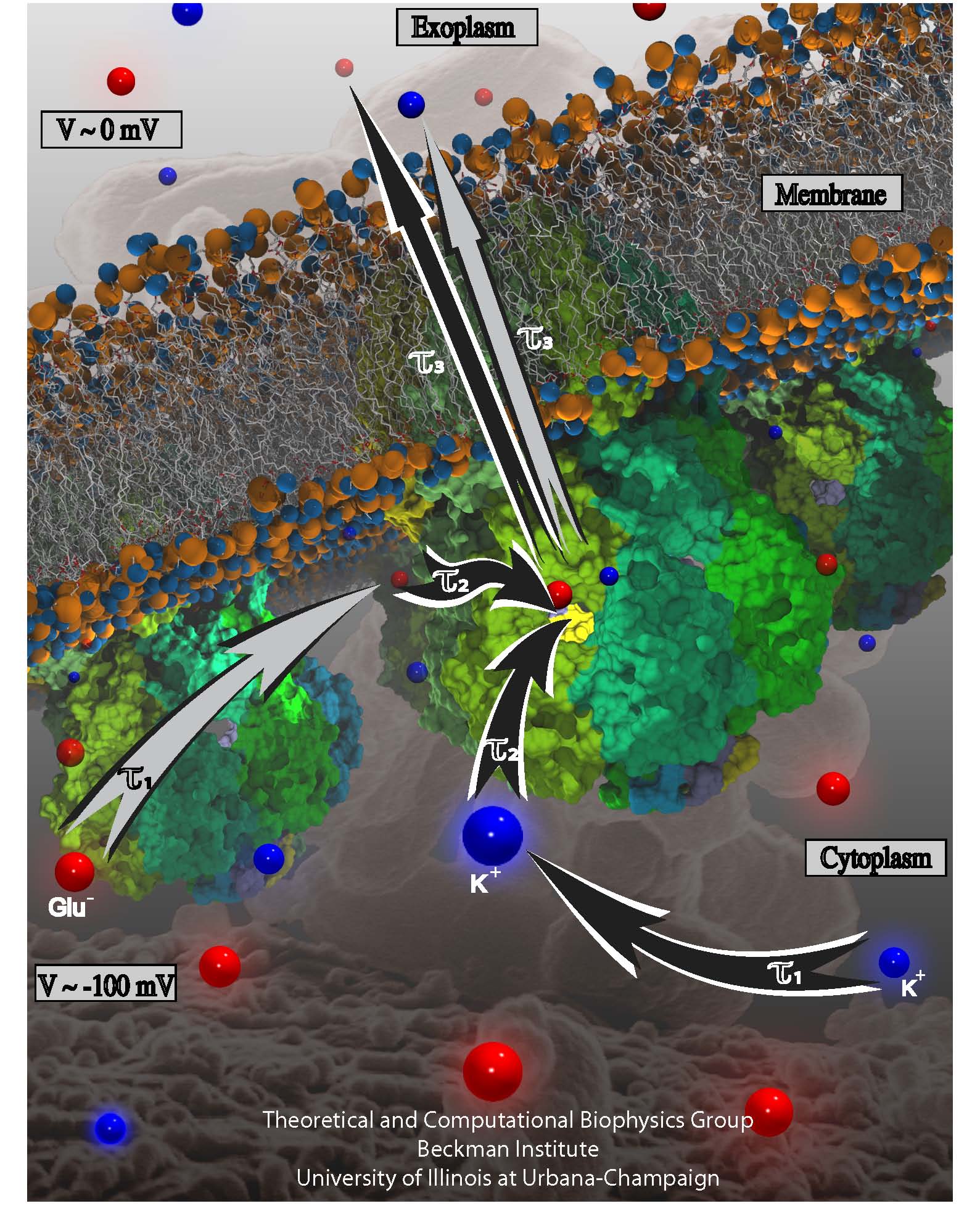
Bacterial cells enclose themselves with a cell membrane to maintain an optimal
interior ion concentration and interior electrical potential. The potential is
about -0.1 V inside versus 0 V outside, the potential difference amounting to an
important energy reservoir for the cell. Closing itself off from the outside
can be dangerous, though, for a cell, namely when sudden changes outside the
cell can bring a cell to burst. This can happen when ions outside the cell are
washed away suddenly: outside water pushes itself then into the cell as the
water prefers to be near ions found then only inside the cell, a behavior called
osmosis. The osmotic push can be so hard that the cell can burst. To prevent
such burst, cells evolved safety valves, one being called mechanosensitive
channel of small conductance or MscS. Under pressure, the valve, i.e., MscS,
opens and enough ions leave the cell to keep water from pushing in too hard
(see the Mar 2008 highlight,
"Observation and Simulation depict Cell's Safety Valve",
Feb 2007 highlight, "Observing
and Modeling a crucial Membrane Channel",
May 2006 highlight,
"Electrical Safety Valve", and the
Nov 2004
highlight, "Japanese Lantern Protein").
But since the electrical potential inside is negative,
mainly negative ions would leave the cell, discharging its potential and leaving
the cell without energy.
A theoretical and computational study, the latter
carried out using
NAMD, reports now that MscS developed apparently an ingenious
solution: ions going through the MscS valve must pass a balloon-like filter that
manages to mix positive and negative ions so that only a 1:1 mixture leaves a
cell under osmotic shock, thereby providing protection against the shock without
compromising the cell's electrical potential. More information can be found on
our MscS website.
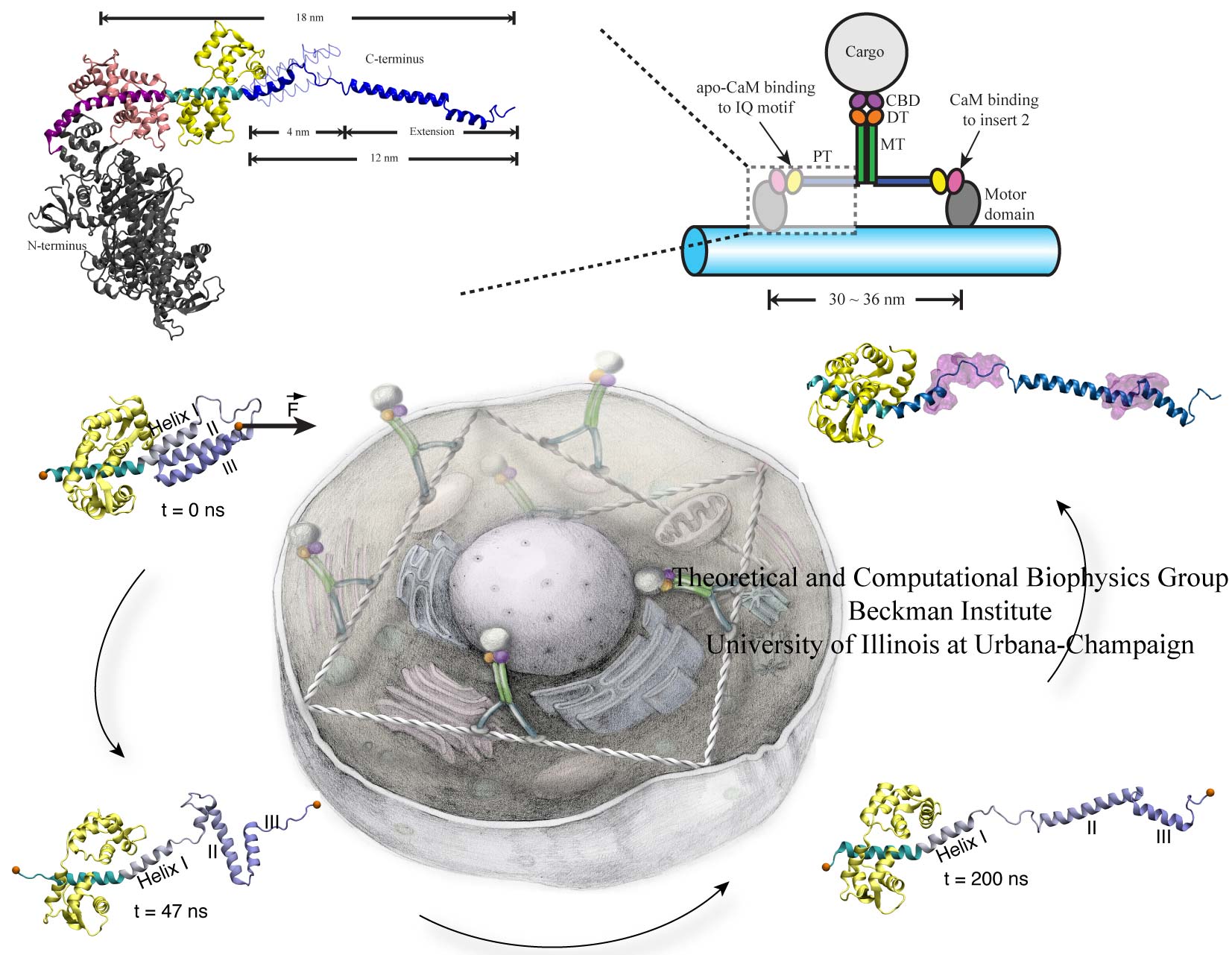
image size:
266.3KB
Motor proteins transport cargos from one place in living cells to another, for example transport cell components along the long axons of nerve cells. A motor protein, such as myosin VI, has to "walk" or "run" along the cellular highway of actin filaments to perform the transport. In the case of myosin VI, snapshots from crystallography revealed that the protein's "legs" are too short to explain the step size taken. Computational and experimental biophysicists have now solved the mystery of how myosin VI dimers realize their large step size despite their short legs.
The investigation, based on the program NAMD and reported recently, demonstrates that the answer lies in the flexibility of the legs. Myosin VI is able to triple the length of each leg, made of a short bundle of up-down-up connected alpha-helices, by extending the bundle to a stretched-out down-down-down geometry of segments, like turning a letter z into a a single long line. In the telescoping process, myosin VI also gets help from its well-known binding partners, namely calmodulins. The calmodulins direct the telescoping of the protein legs as well as strengthen the extended legs. Together with an earlier study of the "neck" region of the molecule (see December 2010 highlight on Opposites Attract in a Motor Protein), the scientists have established how walking myosin VI achieves its wide stride. More information can be found on our motor protein website.
molecular dynamics flexible fitting (MDFF)
to ..." />
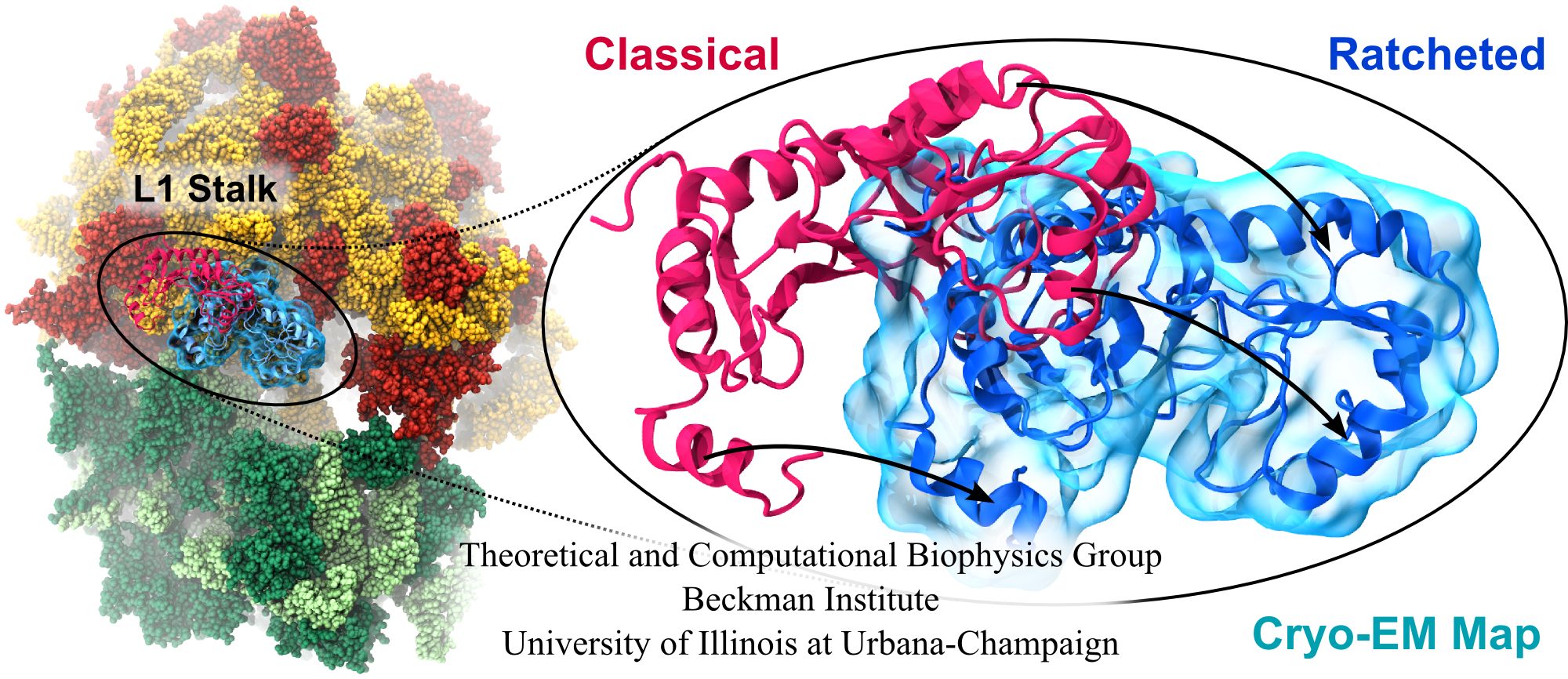
image size:
347.6KB
made with VMD
Water is the essential solvent that shapes protein structure and function,
but for researchers using
molecular dynamics flexible fitting (MDFF)
to fit large biomolecular models to data from cryo-electron microscopy, such as fitting the classical ribosome into the ratcheted map, it was a mixed blessing.
Since the network of hydrogen bonds that gives water its unique properties
must rearrange as the solute moves, water molecules not only increase
the size of the simulation but slow the fitting process.
Leaving water out completely was a common practice, relying on the
MDFF fitting potential to prevent the dehydrated protein from shriveling.
The 2.8 release
of NAMD
provides a better option: a new implementation of the
generalized Born implicit solvent model that scales to thousands of cores
for large biomolecular aggregates thanks to NAMD's unique parallel structure
and measurement-based load balancing system.
By eliminating explicit water molecules from the simulation, an
implicit solvent model helps shape protein structure while adapting
immediately to new conformations.
With this best-of-both-worlds option now available in NAMD,
biomedical researchers using MDFF need fear water no longer.
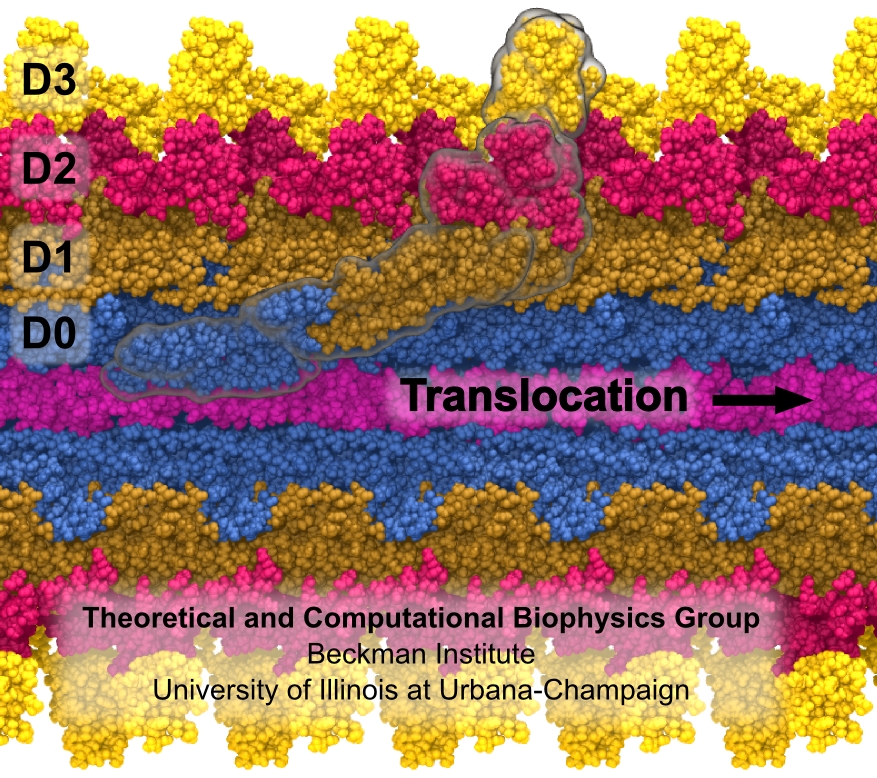
Bacterial cells can swim and use for this purpose one or more flagella, whiplike appendages that exceed the length of the cell severalfold.
The flagella are made of many thousand copies of a protein called flagellin, arranged in a helical fashion such that the flagella are hollow inside, forming a very long channel.
When the flagella are rotated by the cell counter-clockwise, the cell swims straight; when they are rotated clockwise, the cell turns to a new direction.
Through swimming and turning the cell searches its habitat for food and avoids trouble.
But sometimes a flagellum breaks and needs to grow back.
At this point starts an amazing process: the cell makes new flagellin and pumps the unfolded protein into the flagellar channel, extending its length.
This is like squeezing toothpaste out of a tube, except in reverse, like pumping toothpaste into the tube at the toothpaste factory, and the tube is extremely long.
Now researchers have described the process that makes flagella grow step-by-step through a combination of mathematics, physics, and molecular modeling using NAMD.
As reported, the researchers reproduce the time course of growth as well as the length of the growth and also explain how friction of the protein paste is kept extremely low to make the flagella grow many times the length of the cell itself.
More information here.
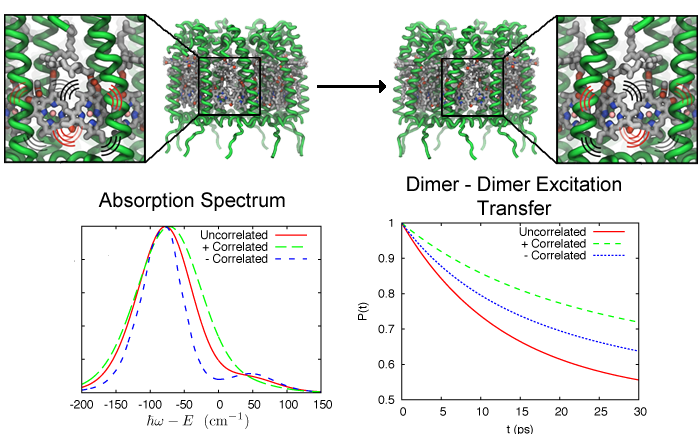
image size:
231.6KB
made with VMD
Photosynthetic life forms bottle the energy of sun light. How do they
do it? Fast! Indeed, the first nanosecond after absorption of sun
light is crucial in photosynthetic light harvesting. During this time
absorbed solar energy is in its least stable form, that of
electronically excited molecules which decay by re-emitting a photon
(fluorescence) at a rate of 1 every nanosecond (1 every 0.000000001
seconds). Fluorescence would be wasteful to the organism and to avoid it
the molecular excitation energy is transported over tens to hundreds of
nanometers through an energy transfer network to so-called
photosynthetic reaction centers where it is converted into a more stable
form of energy (see our recent review on light harvesting). The fast
transport is achieved by transferring excitation energy between clusters
of strongly interacting pigment molecules that act as stepping stones
and as a result the excitation energy is used in about 0.1 nanoseconds,
i.e., within 10% of the fluorescence decay time, thus bottling sun light with an
efficiency of 90%. The thermal motion of the pigment molecules and their
protein scaffold greatly influences the excitation transport. A recent study
showed that correlated thermal fluctuations that arise in pigment
clusters affect the excitation transfer particularly strongly, typically
slowing transfer it down. Pigment clusters that avoid correlated thermal
motion increase the efficiency of light harvesting. More information can
be found here.
VMD
has evolved to version 1.9, featuring
new and improved tools for molecular cell biology.
A ..." />


image size:
1.9MB
Olga Svinarski and VMD
The molecular graphics program
VMD
has evolved to version 1.9, featuring
new and improved tools for molecular cell biology.
A key new tool in VMD permits visual diagnosis of the long-time dynamics
of large structures.
Improved computer power permits now simulation of processes like protein
folding that stretch over microseconds to milliseconds.
While short in human time, the process measured in terms of basic
molecular motion like bond vibration is seemingly endless,
involving hundreds of gigabytes of data and long hours of human attention,
if not automated.
VMD offers now with the
Timeline tool,
a convenient way of scanning such data for key "events" that signal
successful process steps.
Timeline can likewise assist in monitoring the dynamics of large
cellular machines involving millions of atoms.
VMD 1.9 sports other new tools like
ParseFEP
for analyzing so-called free energy perturbation calculations determining
the energy arising in reaction processes as calculated with VMD's sister
program NAMD.
VMD includes now also a lightening fast tool to detect
spatial regularities in the arrangement of small molecules in order to
detect if they constitute disordered or ordered arrangements. By
calculating the so-called
radial distribution function
the tool could
identify plug formation in nanopore sensors due to precipitates that go
undetected by density or visual inspection. Pleasing to the eye, molecular
graphics features in VMD have been enhanced, enabling
faster ray tracing,
new shading features,
X3D
molecular scene export for display in
WebGL-capable
browsers including Chrome, Firefox, and Safari.
See more on the VMD 1.9 release page.
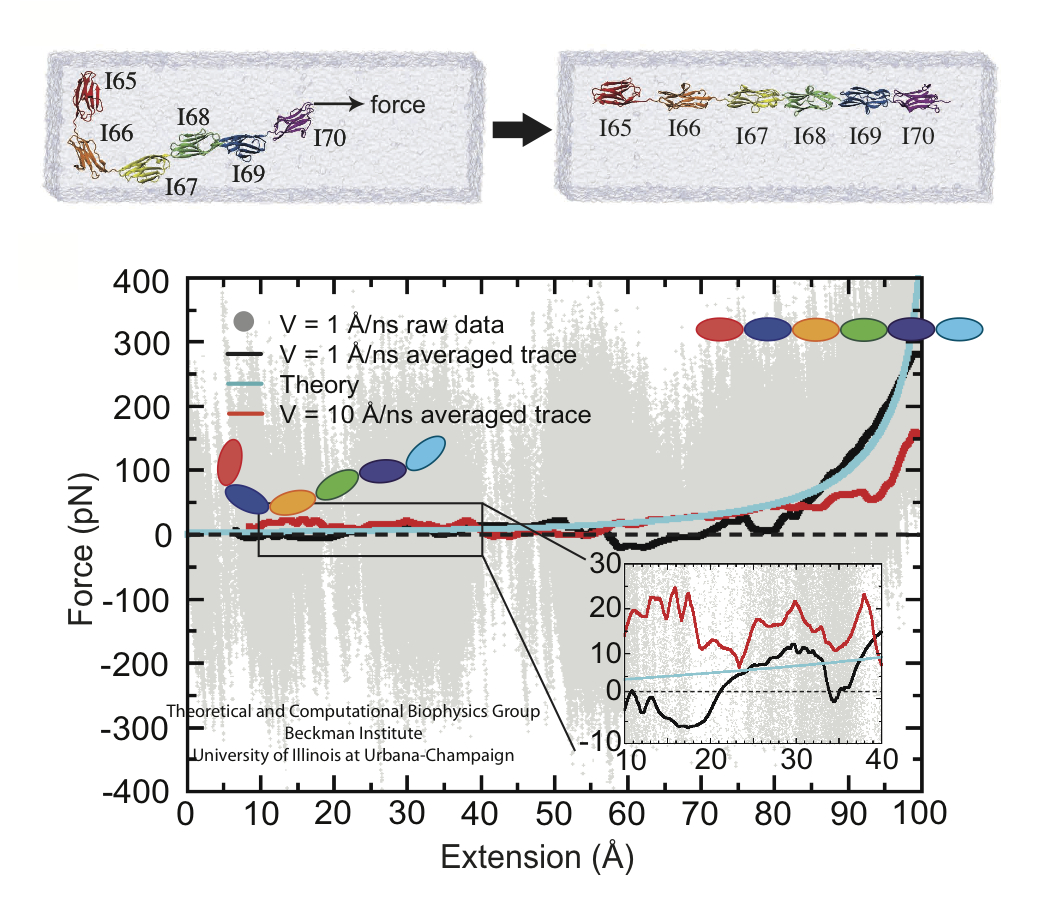
image size:
60.3KB
made with VMD
A cell that is alive can be recognized typically under a microscope through cell motion, streaming of inner cell fluids, and many other mechanical cell activities. These activities are the result of mechanical forces that arise in the cell, but even though the effect of the forces on cells are tremendous, the magnitude of the forces when measured by physical instruments, are extremely small, 1/1000000000000 of the force felt when lifting a Kilogramm. Sensing such small force requires extremely high precision in research techniques, and, for more than a decade, computational biologists, using NAMD, have exploited the all-atom resolution of molecular dynamics modeling to explain very successfully the molecular level effect of small forces in cells (for example, see our previous highlights on muscle protein, blood clot protein, and other successful applications). But simulations, too, are challenged by the small magnitude of cellular forces, mainly because precise simulation requires extremely extensive simulations, that cover a duration as close as possible to the biological time scale. Advances in computational biology have now permitted in the case of a the muscle protein titin a simulation of unprecedented accuracy that resolved clearly the relationship between molecular structure of titin and its ability to sustain the forces that arise in muscle function. For this purpose a key element of titin was stretched at a velocity slow enough that hydrodynamic drag, that came about as an unwanted byproduct of earlier simulations, was negligible compared to titin's intrinsic force bearing properties. The new simulations opened an unveiled view on muscle elasticity. More information can be found on our titin website, and in a recent review .

image size:
118.5KB
made with VMD
movie (YouTube)
Molecular modeling simulates the motion of cellular biomolecules at the
atomic level. To make the simulations faithful, the physical forces
acting between atoms need to be described accurately. Electric field effects
between atoms, so-called atomic polarizabilities, are especially difficult
to model well in a computationally cost effective way. There is an ongoing
effort in the molecular modeling community to develop cost effective models
that more faithfully represent the microscopic properties of biomolecules
due to the ambient electric field effects. Recent development work has added
support in the simulation program
NAMD
for one of these advanced modeling efforts.
As reported,
the new algorithms used in NAMD achieve good parallel computing performance,
with a cost that is not more than twice that of the standard model,
not accounting for atomic polarizabilities. The new model
is demonstrated to reproduce many physical properties better than the
standard model, including more accurate bulk density and surface tension
at the interface between liquids and more accurate diffusive behavior of
ions in a solution. More on our
research webpage.
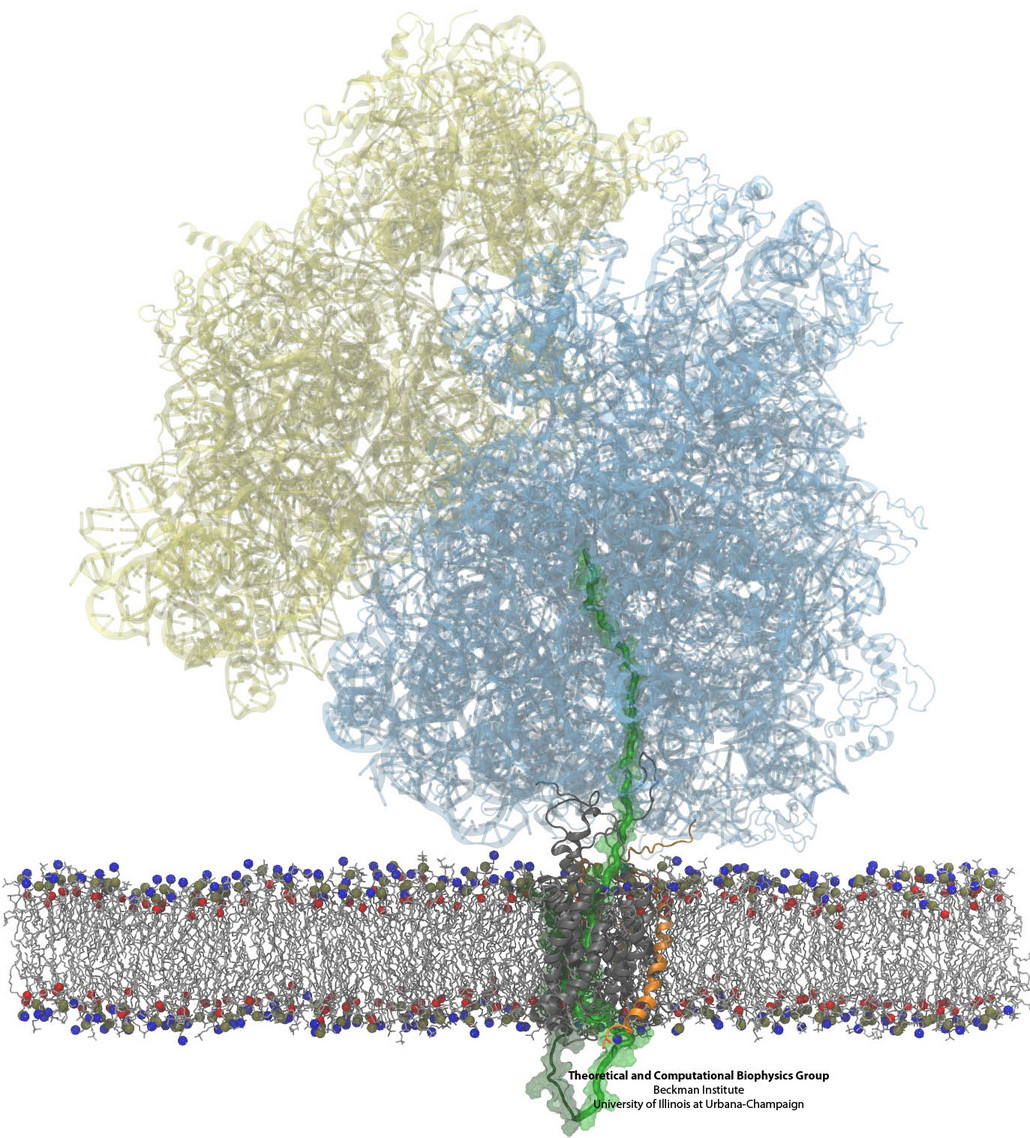
Constructing and correctly placing new proteins is a complicated task for living cells. Starting with nothing more than a sequence of DNA, the cell has to
translate the genetic code, stitch together the constituent amino acids, and then place the newly made protein where its function is needed. To accomplish these
feats, the cell using tools such as the ribosome, a protein factory (see the Dec. 2009 highlight Managing
the Protein Assembly Line), and the protein-conducting
channel, a switching station within the membrane (see the Nov. 2008 highlight Patching a Leaky Channel). All
instructions for making a nascent protein and localizing it, e.g., to the watery
cytoplasm or the oily membrane, are carried within its DNA sequence, but how it is read and then executed has long been unclear. Now, two recent research
advances picture these processes in astonishing detail. The first advance (reported here), from a collaboration between cryo-electron microscopists and
computational biologists using MDFF, shows an atomic level structure that caught the ribosome in the act of inserting a protein into a membrane. The structure
reveals the newly forming protein transiting from within the ribosome to the channel and then through an open gate in the protein-conducting channel into the
membrane. The second advance (reported here), accomplished with the simulation program NAMD, explained how the
ribosome and the protein-conducting channel manage to pay the energy price of inserting a new protein, one
amino acid a time, into the membrane.
For more information see our protein translocation website.
review
explores how the Graphics Processing Units (GPUs) found in commodity
high-end video cards are increasingly
being used not ..." />
A recent review
explores how the Graphics Processing Units (GPUs) found in commodity
high-end video cards are increasingly
being used not only for interactive molecular graphics,
but also for molecular simulation and analysis.
NAMD and
VMD both support GPU-acceleration
using NVIDIA CUDA, enabling computationally demanding
simulation, visualization and analysis tasks (e.g., of the
electrostatics of Tamiflu binding)
to be run with shorter
turnaround on modestly priced GPU clusters, desktop, and laptop computers.
This affordable computational power is particularly compelling for
interactive modeling, with a
recent report
detailing how interactive molecular dynamics simulations with haptic feedback
are now possible on GPU-accelerated desktop computers.
Three recent book chapters detail the application
of GPU computing techniques to the calculation of
electrostatic potentials,
interactive display of molecular orbitals,
and more general
molecular modeling algorithms.
In August 2010 the Resource held its first
workshop on GPU programming
for molecular modeling
to bring the benefits of GPU computing to a broader range
of molecular modeling tools and magnify the impact of our
GPU computing research.


