Highlights of our Work
2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
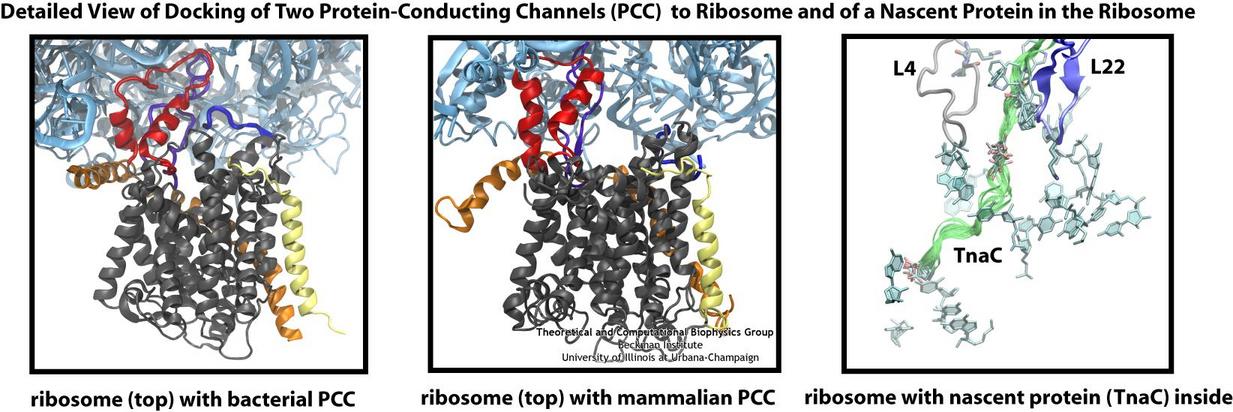
Living cells contain millions of proteins composed of sequences of different amino acids that typically fold spontaneously into well-defined three-dimensional conformations and then carry out
their role as molecular machines serving manifold functions in the cells (see the protein folding highlight). The synthesis of the
proteins is carried out by the ribosome, one of the largest molecular machines present in all cells, which reads the cell's genetic information for the purpose. Three researchers were
recently awarded the 2009 Nobel Prize in Chemistry for the determination of the ribosome's structure. The physical
mechanism of the ribosome, the cell's protein factory, is still largely unknown. Just as in any factory, there are multiple directions and controls on the protein assembly line. Sometimes the
protein products need to be redirected to different parts of the cell, and other times assembly needs to be halted altogether. Now, significant new insights into both of these aspects of
protein assembly have been made by combining electron microscopy with molecular dynamics simulations using the recently developed molecular dynamics flexible fitting method (MDFF, see the June 2008 highlight). This combination allowed researchers to visualize the complex between
the ribosome and a protein-conducting channel that directs proteins into and across membranes for both a mammalian system (reported here) and a bacterial system (reported here). Amazingly, despite the
evolutionary distance between mammals and bacteria, both complexes are remarkably similar. Simulations of the bacterial ribosome-channel complex, among the largest ever performed, further
revealed the steps in the direction process. In a third study (reported here), researchers determined how TnaC, as a protein newly
synthesized by the ribosome, can stall the ribosome from within during its own assembly, which then controls the expression of related genes. More information on these unique protein assembly
controls can be found on our ribosome and our protein-conducting channel websites.
image size:
268.6KB
made with VMD
Nanotechnology is flourishing today, for example building entire chemistry laboratories on a small chip. In doing so nanoengineers experiment with different materials, one of the latest being good old plastic, chemically known as polyethylene terephthalate, or PET. Presently, nanometer-size pores, so-called nanopores, are built from silicates and PET membranes. To image how nanopores filter ions (see the Mar 2009 highlight) or affect DNA mechanics (see the Feb 2009 highlight) one can employ molecular dynamics (MD) simulations using NAMD. Such simulations provide a realistic representation of the devices, which can be employed to improve the technical uses of nanopores. In a recent publication, MD simulations revealed that the conduction properties of PET nanopores are controlled by chemical properties of the inner surfaces of the nanopores, for example, if surface groups sticking into the nanopore are protonated or not. The studies show once more the value of molecular dynamics simulations as computational microscopes, providing to nanoengineers the atomic-level visualization of extremely small devices. More information is available on our PET nanopore website.
Theoretical ..." />


image size: 493.4KB
Twenty years ago the Beckman Institute opened on the campus of the University of Illinois and the Theoretical and Computational Biophysics Group (TCBG) was the first to move into the beautiful new building. The group advanced computational biology through widely used software and exciting discoveries (some recently described here). Hence, it was time to celebrate and so TCBG did with the symposium Computational Biology of the Cell - The Next Decade. But rather than looking backward, the symposium glanced into the future that promises as many great opportunities as the last two decades realized: computational power for millisecond atomic resolution simulations, coarse-graining methods to describe true cellular size and times scales, better force fields, closer collaboration with experiment, and partnership with nanotechnology. Bringing together many of today's leading thinkers and practitioners of biomolecular modeling lectures led to lively debates (see some photos) regarding computational biology's future. On TCBG's past and future read more here.
Proteins are molecular machines inside living cells which carry out tasks from chemical synthesis to cellular motion. Each protein is a linear polymers
made of units that each are one of twenty amino acids, A(1) to A(20),
for example from a sequence A(5) - A(12) - A(1) - A(5) - ... - A(17).
Random sequences lead to disordered, non-functional proteins.
A central feature of life is that evolution has given rise to
protein sequences which fold into functional proteins. The
process through which the disordered linear polymer folds
into its final structure, however, remains mysterious, since it
is very difficult to observe experimentally. Unfortunately,
simulations of protein folding are very computationally
demanding and have uncertain outcomes (see the May
2009 highlight), which has historically
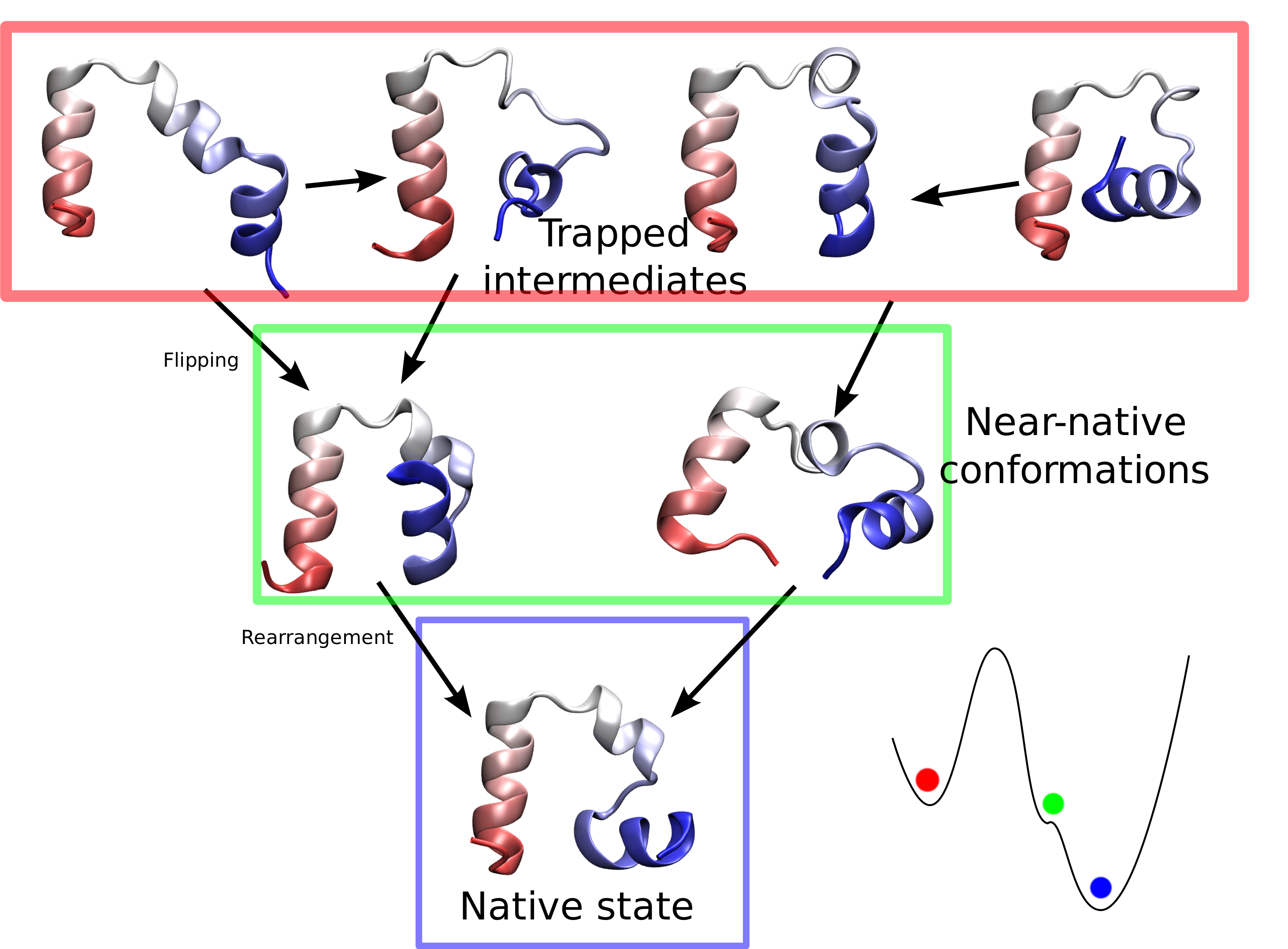
limited their utility. Now, researchers have used NAMD to simulate the
complete folding process of a small protein, villin headpiece, as reported
recently. In a series of atomic resolution molecular dynamics
simulations, covering a total of 50 microseconds, multiple
folding events were observed. Importantly, the simulations
provided a glimpse at the prevalent intermediate conformations
visited by villin during folding. A key transition between two
intermediates was recognized as rate-limiting in the villin
folding process. More information is available at our protein folding
website.
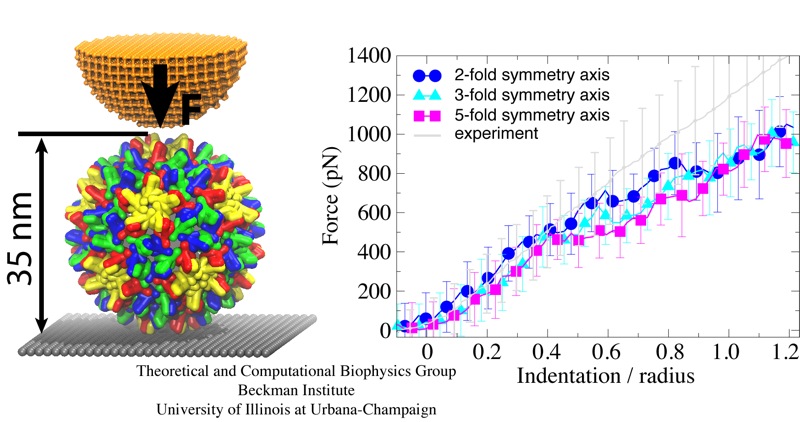
Viruses are the simplest life forms known. In fact, one can question if they are life forms at all, as they cannot exist without infecting a host cell and using its machinery for replication. The virus is indeed just a package material surrounding a genetic message that instructs the host cell to replicate the virus. It looks like a soccer ball, but is
a million times smaller
(see also the March 2006 and January 2007 highlights). The infection, a well known example being infection of human cells by a flu virus, involves the virus to approach a human cell and dock onto it, become internalized by the cell, bursting then its package, called the capsid, and release the genetic message. The virus capsid needs to be sturdy and impermeable up to the approach to the cell, but then become brittle and porous to release the genetic material. Obviously, the virus capsid must have very distinct mechanical properties to function. To investigate these properties experimental and computational biophysicists teamed up. The experimentalists placed empty capsids of the hepatitis B virus onto a small chip and mechanically squeezed the capsid then with an extremely small tip, measuring how much force is needed to squeeze the spherical capsid down repeatedly. Computational researchers using NAMD repeated the experiment in simulation. As they reported recently, simulation gave the same forces as the experiment, but yielded also a detailed picture of the capsid mechanics. More on our "Molecular dynamics of viruses" web site.
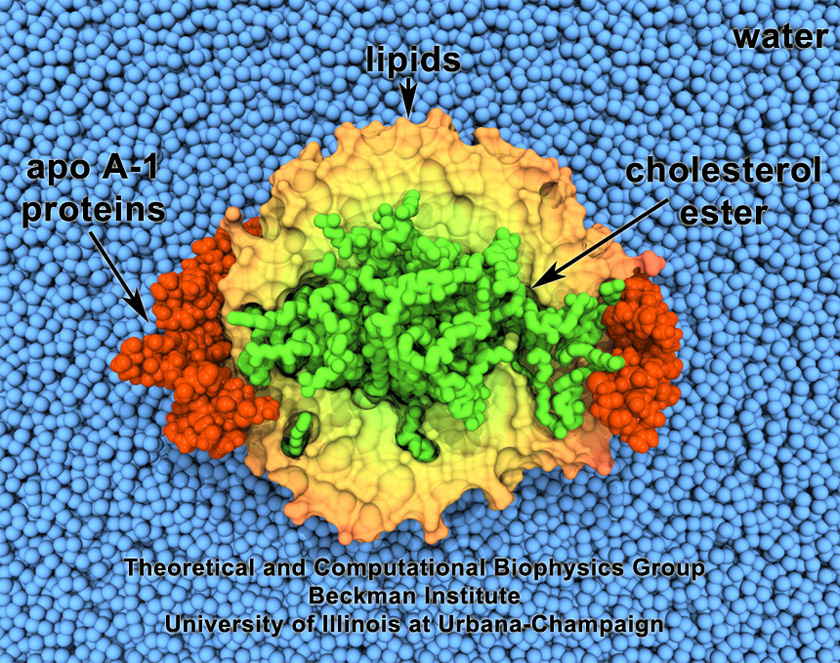
image size:
496.6KB
made with VMD
Cholesterol maintains a healthy body, but too much cholesterol can lead to atherosclerosis
and heart disease. Lipoproteins can bag superfluous cholesterol in the arteries and
transport it to the liver for removal. One such lipoprotein is high-density lipoprotein
(HDL) which self-assembles into discoidal particles (see the Mar 2007
highlight) and then bags cholesterol. How this works is the subject of a recent
report. Molecular dynamics simulations using NAMD revealed that discoidal HDL
particles, teaming up with the enzyme LCAT, first turn cholesterol chemically into
cholesterol ester and then sucks it into the interior of the particle; in the course of
this process, the HDL particle swells into a sphere. More information on our lipoprotein website.
image size:
440.0KB
made with VMD
Proteins are the workers in cells; they carry out designated cellular functions tirelessly throughout their lifetimes. Some proteins can even hold two different jobs. One example of a dual-duty protein is the bacterial photosynthetic core complex. The photosyntehtic core complex performs the first steps of photosynthesis: absorption of sunlight and processing of light energy. Besides providing solar power, the core complex acts as an architect of the cell by shaping membranes in the interior of photosynthetic bacteria. Combining computational modeling and electron microscopy data using the Molecular Dynamics Flexible Fitting method, computational biologists have recently reported studies of both functions of the core complex, namely, the light-absorbing features and the membrane-sculpting properties. More details can be found on our photosynthetic chromatophore website.
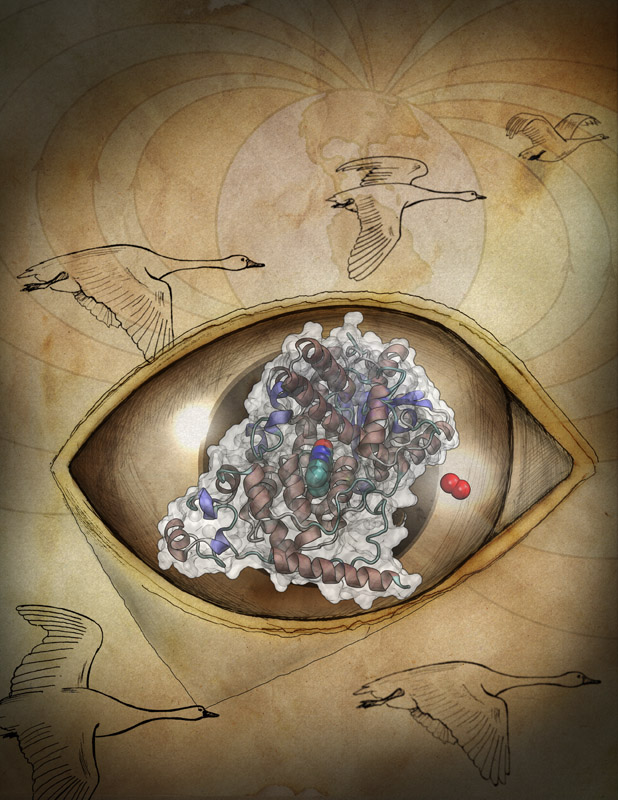
image size:
181.3KB
made with VMD
Many animals have a magnetic sense that tells them North from South. Migratory birds, salmon, or sea turtles migrate thousands of miles relying on this sense, but also animals staying closer to home like honeybees, newts or lobsters use it. Most likely, the animal magnetic sense is based on two types of receptors, one based on magnetite, another one on a protein called cryptochrome found in animal eyes, including human eyes. So why do we humans not enjoy the magnetic sense? A recent report, offers a very intriguing answer. In sensing the Earth's magnetic field, cryoptochrome relies on so-called redox reactions which exchange electrons between molecules. Such reactions are crucial for life, but can also be damaging; antioxidants are used by organisms, but also in pharmacology and as dietary supplements to keep the reactions in check. Apparently, cryptochrome recruits as a reaction partner in its magnetosensitive behavior a special form of molecular oxygen, namely its negatively charged brethren superoxide. For this purpose cryptochrome requires superoxide in low doses, which is good since superoxide, though arising in organisms and used in signaling elsewhere in the body, is actually toxic. The human body has an extremely efficient enzyme, superoxid dismutase, that keeps superoxid at a very low concentration level, apparently too low for human cryptochromes to capture it and tell North from South. Humans, somewhere in evolution, might have lost the magnetic sense, but gained longevity. More on our cryptochrome web site.
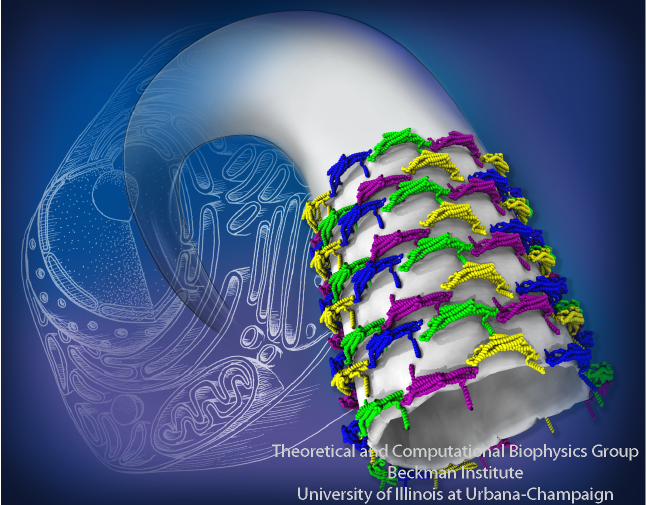
Living cells are characterized by a great diversity of separate internal spaces, the boundaries of which are made of membranes forming convoluted surfaces of manifold shapes. Sculpting these shapes is achieved in many cases by proteins. A single protein is too small to bend the membrane into useful shapes, such as spheres or tubes, that measure 10-100 nm, or more, in diameter. Indeed, the proteins work in teams, but exactly how remained a mystery. Now a computational study elucidates the membrane-sculpting process for proteins called amphiphysin N-BAR domain. Simulations performed with NAMD had revealed a first glimpse earlier (see the Sep 2008
highlight). The new study showed that multiple N-BAR domains form lattices maintained through electrostatic interactions. Positively charged, banana-shaped surfaces of individual proteins bend the negatively charged membrane, while the lattice formation ensures a uniform bending force across a wide membrane surface. In a dramatic example of computational "microscopy" the 200 microsecond sculpting of a large flat membrane into a complete tube was observed. More here.
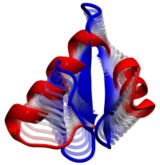
Computational modeling seeks to simulate biomolecules, particularly
proteins. The dream of computational biologists is that their
simulations realistically mirror the structure and dynamics of
proteins, which act as molecular machines in living cells. Indeed,
when researchers tried to simulate how a nascent protein folds into
its known shape the chosen protein, called WW domain, did not fold
properly (see the May 2008 Highlight).
Until recently, simulations could follow protein movement
for about 0.0000001 seconds and the computational mirror seemed to work
well. Recently, however, simulations with the program NAMD began to
follow proteins for almost 0.0001 seconds, a thousand times longer,
and the mirror showed cracks.
Thus, a question arose as to what went wrong and how the distortion could be repaired. It was unclear whether the simulations still did not last long enough, or whether the physical interactions in the protein were poorly described in the computer model that was used. As reported in a recent
paper, the interactions show subtle errors, significant enough to throw off the energy balance in the folding protein. Fortunately, the results suggest ways to improve the computation of physical interactions to fold proteins more accurately, repairing the cracks in the mirror. More information is available at our protein folding website.
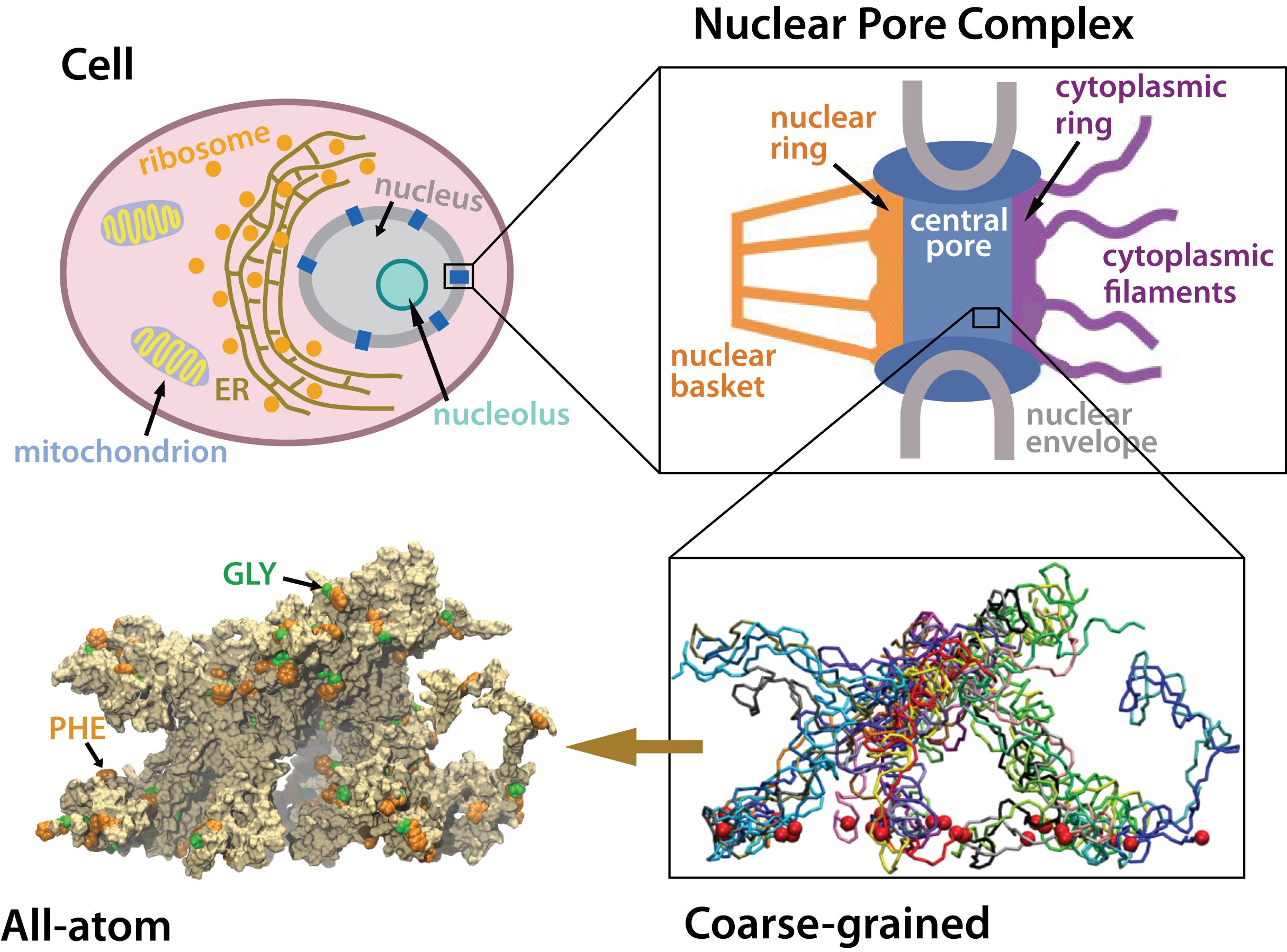
Many living cells, so-called eukaryotic ones, organize their genetic materials
in the cell's nucleus, enveloped by a double membrane with guarded access
through pores that involve an amazing filter. Like an ordinary filter it
permits passage of small particles (biomolecules), but not of large particles
(e.g., proteins). However, certain large particles, proteins called transport
receptors, can pass. The filter is made of long "finger" proteins anchored
inside the pores. The transport receptors can intermittently widen the
filter. But to observe how this is achieved is difficult since the finger proteins
are highly disordered. As
reported recently,
simulations using NAMD suggest now a simple and
elegant answer: the finger proteins bundle in groups of 2 - 6 and form a brush,
filling with its bristles the nuclear pores. The bristles are bundles of finger
proteins and have two key properties: (i) on their surface they are dotted with
spots of amino acid pairs, phenylalanine and glycine, that are known to interact favorably with transport receptors (see the Aug 2007 highlight, the Feb 2007 highlight, and the Jan 2006 highlight); (ii) the bristles are also interconnected, namely where finger proteins change from one bundle to another bundle, which they do with some frequency.
It appears then that the bristles of the nuclear pore filter form an
energetically favorable environment for transport receptors. A
recent report
of new simulations shows that transport
receptors are pulled into the bristles of the nuclear pore filter. More
information here.
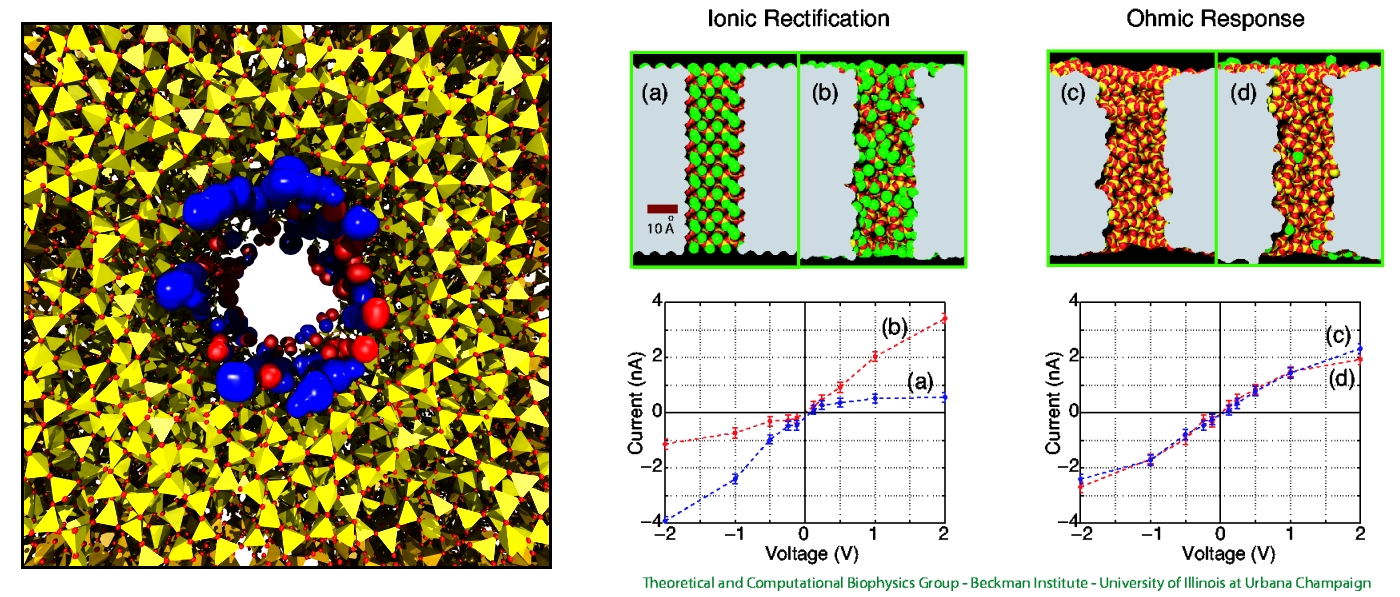
Nanotechnology develops small devices with dimensions below 100 nm, one hundred times smaller than the diameter of a human hair. Nanodevices can be used for a wide range of applications, such as biomedical sensors (see the Jan 2005 highlight) or tools for studying DNA properties (see the Feb 2009 highlight and the Nov 2005 highlight). In building and controlling such small devices, researchers run into problems such as surface effects and significant thermal fluctuations. Furthermore, properties arising from the discrete nature of matter start to dominate at the nanoscale, producing phenomena not observed in larger devices. For instance, when immersed in electrolytic solution and under the influence of an electric field, nanopores act as diodes for ionic currents, conducting in one voltage polarity better than the other, a behavior which has been proposed as the basis for developing nanoelectronic devices. In a recent report, researchers have studied this so-called rectification behavior by means of molecular dynamics simulations using the program NAMD, the ionic rectification inside the nanopore being described in atomic detail. More information can be found here. See also our recent biotechnology review.
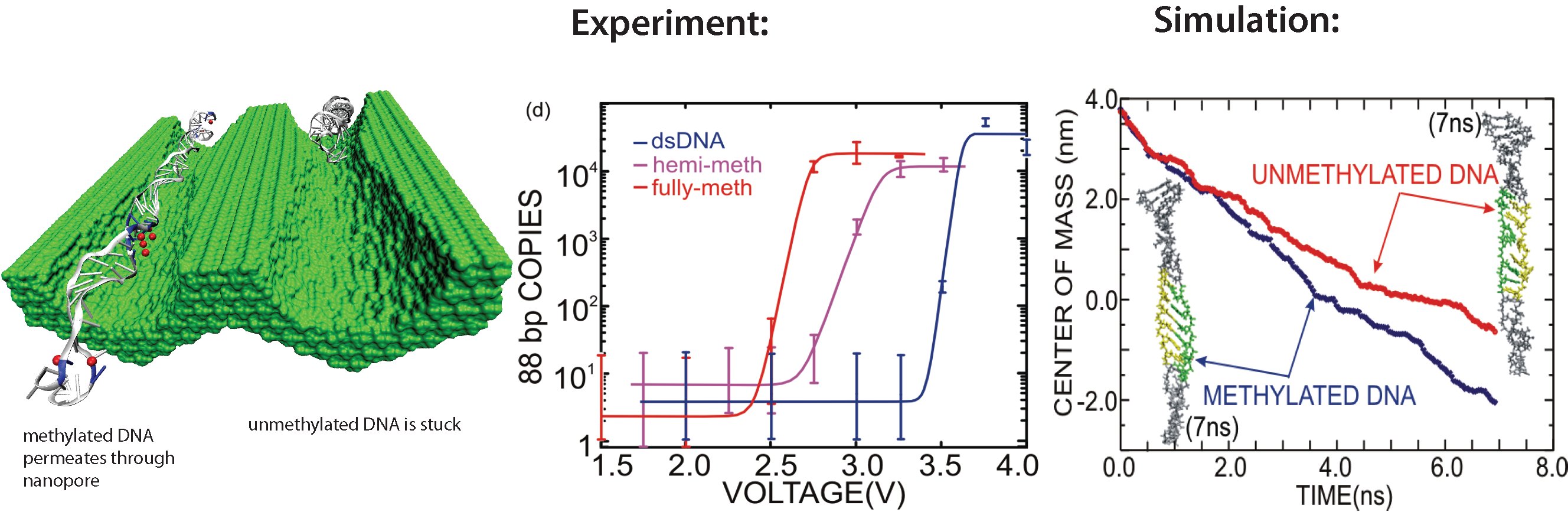
image size:
298.9KB
made with VMD
All cells making up the human body contain the same DNA in their nucleus, the DNA entailing about 30,000 genes and each gene containing instructions for a protein. Despite this sameness, the cells in different parts of our body are very different due to many factors, a key one being that the level of expression of genes into protein is highly regulated and differs strictly from cell to cell. One rather common regulation mechanism involves methylation of one of the four bases of DNA, cytosin. Researchers find that the long DNA in human cells show spots of methylated cytosins, the methylation being correlated with the expression level of the genes near the spots. In fact, medical researcher relate several cancers to improper methylation of DNA. Despite the common occurrence of regulation by methylation, researchers have little understanding how methylation, that changes an H (hydrogen atom) for a CH_3 (methyl group) here and there, i.e., just adds small bumps on a rather bulky DNA molecule, affect the physical properties of DNA such that expression levels are altered. It was found that there are proteins that can recognize the CH_3 groups, i.e., the bumps, on the DNA, but researchers have a hunch that methylation does affect DNA properties directly, i.e., without protein markers, but do not know which properties. In a collaboration between bioengineers measuring the passing of DNA through nanopores and computational biologists simulating this process with NAMD (see also the Nov 2005 highlight stretchable DNA) first hints emerge that methylation does in fact alter DNA's ability to stretch itself through a nanopore. As reported recently, pulling DNA electrostatically through nanopores is easier for methylated than for unmethylated DNA, as seen both in experiment and simulation. The findings promise insight into an important chapter in the field of genetic control. More on our methylated DNA website. See also our recent biotechnology review.
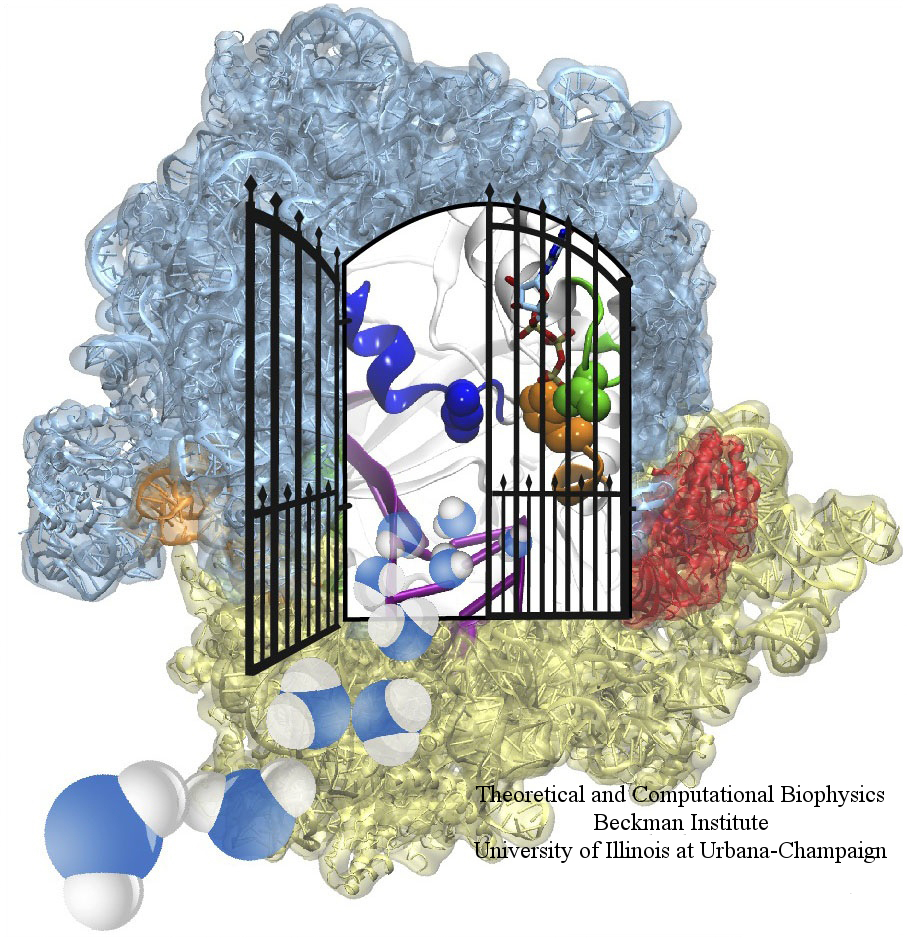
image size: 604.1KB
The ribosome is one of the largest molecular machines present in
hundreds to thousands of copies in every cell, in charge of synthesizing
every protein in the cell faithfully from genetic instruction. For this
purpose the ribosome "reads" the sequence of bases on so-called
messenger RNA, three bases at a time and depending on the base triple,
the codon, elongates a nascent protein by one of 20 possible amino
acids, avoiding to an impressive degree adding a wrong amino acid. So
far one knew that the reading is done by transfer RNA molecules that
have "foots" which match the possible codons and a "head" that brings
along the associated amino acid. Each amino acid has its transfer RNA,
the transfer RNAs checking if the next codon is "theirs," and if it is
they add the proper amino acid to the nascent protein, elongating it.
But how does the ribosome make the critical decision at the decoding
center, namely if the transfer RNA "foot," the so-called anticodon,
matches the codon? The answer is not known, but a key detail has now
been discovered through a combination of electron microscopy and
molecular dynamics simulation using NAMD,
VMD, and a method called flexible
fitting (MDFF, see the June 2008
highlight). It was known that a third molecular system is involved,
called the elongation factor Tu (EF-Tu), which generates a key signal to the
ribosome and transfer RNA through a chemical reaction. This reaction
involves chemically attacking a substrate of EF-Tu, the molecule
guanosine-triphosphate (GTP), with water, breaking a bond and turning GTP
into guanosine-diphosphate (GDP). The puzzle was that EF-Tu is far away
from the decoding center. The collaboration between experiment and
simulation, reported
here, revealed that "correct recognition" through anticodon-codon
binding opens a gate in the EF-Tu that allows water access to the GTP
inducing the signaling reaction. The finding promises to now establish
how the decision at the decoding center is made and how an "open sesame"
order is transmitted to EF-Tu. More on our ribosome website.


