Protein Folding
The ability of proteins to fold into their native state is essential for cell function; misfolded proteins not only lose their function, but can also cause neurodegenerative diseases, including Alzheimer and Huntington. Study of protein folding can aid in preventing protein misfolding diseases and in designing proteins with novel functions. Although most cellular proteins fold on timescales of milliseconds to seconds, several small proteins have been designed and characterized experimentally to fold on a timescale of less than 20 µs. The project will employ Center-developed long-time MD simulation with NAMD to investigate: (1) the folding pathway of λ-repressor and (2) the effect of mutations on folding of the λ-repressor.
Spotlight: Roadmap for Protein Folding (Nov 2013)
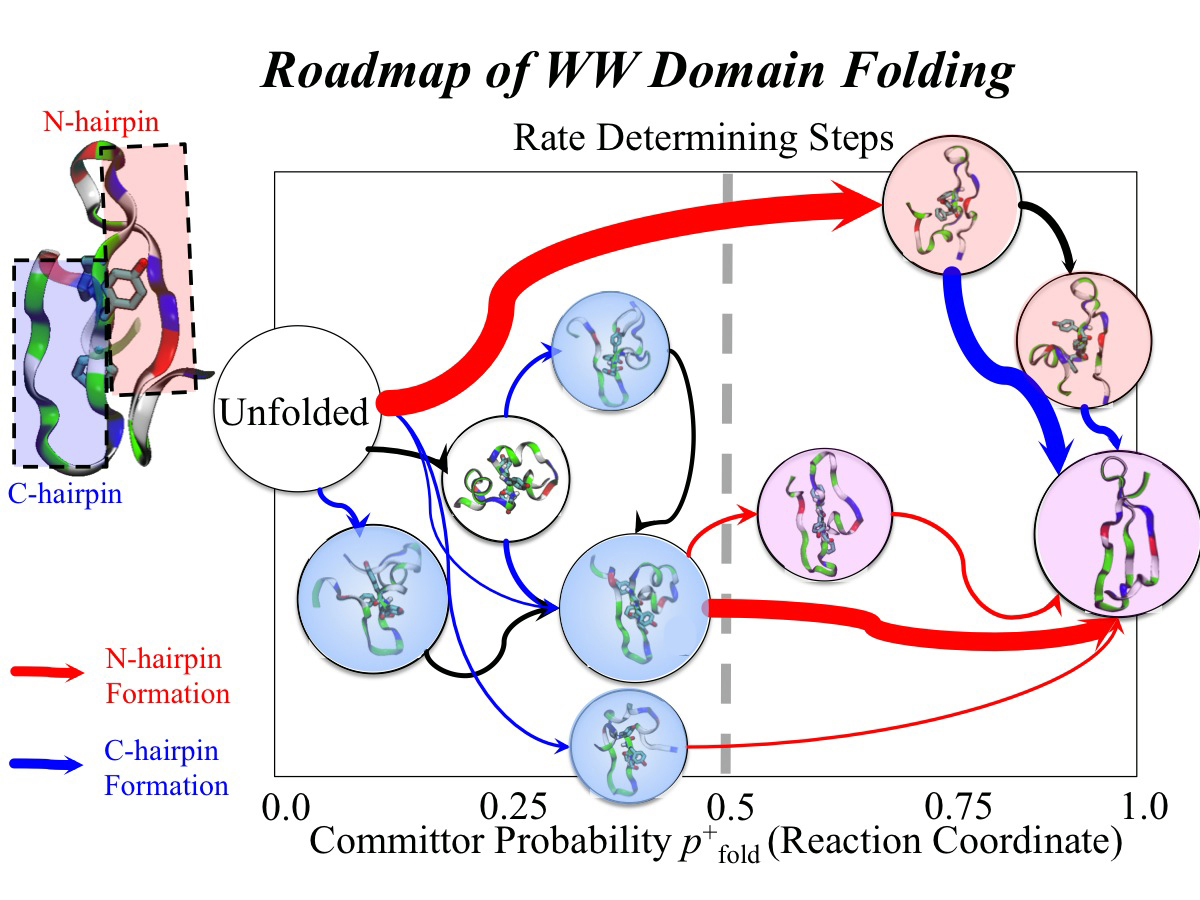
image size:
507.8KB
made with VMD
Every protein in a living cell needs to assume a proper structure in order to fulfill its biological function. The proteins that fails to do so cause malfunction of cells, often leading to disastrous diseases. Thus, how a protein folds into its native structure is one of the central biological processes that need to be understood. The modern view of protein folding suggests that a folding process is governed by structural transitions among a wide spectrum of structures that a protein could access; all of these possible structures and associated transitions constitute a complex roadmap for protein to follow, very much like the one commonly used to navigate a car to its destination through the best route. Obtaining comprehensive and accurate roadmaps for protein folding are essential for the characterization of correct folding routes. In theory, such a roadmap could be explored through computational simulations using an accurate model including every atomistic detail; in practice, the structural complexity of proteins turns the exploration of its roadmap into a daunting computational task. As reported recently, researchers have demonstrated an efficient way to explore roadmaps for protein folding by utilizing a hybrid model which combines atomic models for the protein part with a fast simplified model for the surrounding water part in order to achieve both accuracy and efficiency. By investigating folding mechanisms of two proteins, the researchers showed that their approach is able to generate folding roadmaps as accurate as those obtained with complete atomic models while only taking days to finish the task, much faster than the complete atomic models that usually need months. For details, please see our hybrid-resolution model website.


